Malaltia de Stargardt
La malaltia de Stargardt, també coneguda com a distròfia macular juvenil, és una malaltia ocular hereditària que es caracteritza per una degeneració macular, la màcula és la zona central de la retina dotada de la màxima sensibilitat que permet la visió fina dels detalls.[1] L'inici dels símptomes té lloc en l'adolescència, encara que existeix algun cas que ha tingut manifestacions en la primera dècada de la vida. El pacient refereix que a poc a poc deixa de veure sense adonar-se i es manifesta per perduda d'agudesa visual progressiva.[2][3][3]
| Tipus | degeneració macular associada a l'edat i malaltia |
|---|---|
| Especialitat | oftalmologia |
| Patogènia | |
| Associació genètica | ABCA4, CNGB3, PROM1 i ELOVL4 |
| Classificació | |
| CIM-10 | H35.5 |
| Recursos externs | |
| OMIM | 248200, 600110, 603786 i 248200 |
| DiseasesDB | 31282 |
| MeSH | D000080362 |
| Orphanet | 364055 |
| UMLS CUI | C1858080, C0271093 i C1855465 |
| DOID | DOID:0050817 |
Història modifica
La primera descripció va ser realitzada l'any 1909 per l'oftalmòleg alemany Karl Stargardt. El 1965, Franceschetti li va donar el nom de fundus flavimaculatus. Actualment poden denominar-se indistintament malaltia de Stargardt o fundus flavimaculatus.[4] El 1997, els investigadors van aconseguir aïllar el gen de la distròfia macular de Stargardt: l'ABCA4. Aquest gen produeix una proteïna que exerceix un paper important en el transport d'energia des d'i cap a les cèl·lules fotoreceptores de la retina. Aquesta malaltia deu el seu nom a l'oftalmòleg alemany Karl Stargardt encara que al principi es deia només distròfia macular juvenil. Actualment no existeix un tractament efectiu per combatre la patologia causada per aquest al·lel hereditari, però disposar del coneixement de les bases genètiques de la malaltia pot ajudar a desenvolupar noves estratègies terapèutiques per corregir-la.
Freqüència modifica
Es considera una malaltia rara, es presenta un cas per cada 10.000 persones aproximadament.
Herència modifica
És una malaltia de transmissió hereditària segons un patró autosòmic recessiu. Està provocada per una mutació en el gen ABCA4, que codifica una proteïna que només s'expressa en la retina i que és un transportador de membrana de les cèl·lules fotoreceptores. Es coneixen més de 558 mutacions diferents que poden originar el mal.
Símptomes modifica
Els símptomes més comuns al començament d'aquesta malaltia, sol referir-se el pacient al fet que les coses desapareixen i tornen a aparèixer, que es veuen punts negres que estan i no estan. Això succeeix en els primers estadis i sol ser aproximadament cinc anys abans que existeixi alguna dada que verifiqui aquesta malaltia. La retinografia està neta i el pacient refereix que no veu ni amb la major correcció possible. Cursa en ocasions visió borrosa, zones cegues en el camp visual que es diuen escotomes i dificultat per adaptar-se a les penombres (llocs amb poca llum). Aquelles persones que pateixen la distròfia macular de Stargardt són molt sensibles a la llum i es veuen obligats a usar ulleres fosques (filtres taronges) així com visera o gorra. Han d'evitar la ingesta de vitamina A i prendre el sol durant tot l'any; per a això es posaran crema solar i evitaran estar exposats innecessàriament al sol.
Fons d'ull modifica
En les primeres fases la disminució de l'agudesa visual és clara però no s'observa grans alteracions en el fons d'ull. A mesura que progressa la malaltia, fragments rics en lípids s'acumulen en la capa de l'epiteli pigmentari de la retina per sota de la màcula, els quals apareixen com a taques groguenques. L'epiteli pigmentari de la retina és una capa que es troba entre la retina i la coroides, que és la responsable d'irrigar i nodrir a les cèl·lules fotoreceptores de la retina que són els cons i bastons. Aquests fragments de lípids es diuen lipofuscina. En els casos avançat de la malaltia, aquesta acumulació progressiva de lipofuscina provoca l'atròfia de la màcula i de l'epiteli pigmentari de la retina.
Característiques clíniques modifica
Existeix una àmplia varietat de troballes clíniques:
- Màcula vermelló per epiteli pigmentari retinià molt pigmentat
- El més característic són taques vermell-groguenques (flecks) per acumulació de lipofucsina a nivell de l'epiteli pigmentari retinal (EPR) que varien en forma, grandària i distribució
- Signes: La fòvea pot ser normal o tenir un clapejat inespecífic; l'atròfia geogràfica pot tenir una configuració en ull de bou; la lesió foveal ovalada té un aspecte de "bava de caragol" o "bronze colpejat" en alguns casos envoltada de punts blanc-groc.
-
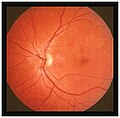 Estadis inicials
Estadis inicials -
 Estadis inicials
Estadis inicials -
 Estadi avançat
Estadi avançat -
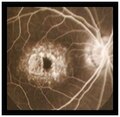 Fluoresceinografia digital
Fluoresceinografia digital -
 Microscòpia electrònica d'escombratge
Microscòpia electrònica d'escombratge





Tractaments modifica
Encara no existeix tractament disponible per a la Malaltia de Stargardt. Inclusivament així, els següents tractaments estan en fase experimental.
- Teràpia gènica: aportació d'una còpia no mutant del gen afectat mitjançant un vector de transferència gènica viral portador del gen ABCA4 normal. (Stargen)
- Trasplantament de cèl·lules pigmentaries a partir de cèl·lules mare embrionàries (Advanced Cell Technology)
- Trasplantament de retina a partir de copes embrionàries generades in vitro (Sasai)
- Fenretinide
- Xips robòtics Argus II
Exàmens complementaris modifica
- Campimetria que mostra l'existència d'escotomes.
- Estudis elèctrics de retina: electrorretinografia i electrooculograma, poden donar patrons variables fins i tot normals.
- Angiografia amb fluoresceina: mostra una coroides fosca a causa de dipòsits de lipofuscina dins de l'epiteli pigmentari de la retina i hiperfluorescència macular a causa de l'efecte finestra.
- Seqüenciació genètica per determinar la mutació de la qual és portador el pacient. És molt important i és la prova definitiva per poder saber quin ha estat la mutació i en quina seqüència s'ha produït. Ara no és important però ho serà una vegada que es desenvolupin més assajos clínics amb èxit. És recomanable que tots els pacients que tinguin dubtes o que el seu oftalmòleg els digui que esperin, que sol·licitin aquesta prova ja que amb una anàlisi de sang es pot determinar en aproximadament sis mesos.
Ajudes òptiques modifica
Els pacients tenen baixa visió i no es corregeix amb cap tipus d'ullera, per tant han d'utilitzar ajudes òptiques que els permeten realitzar les activitats diàries.
- Ulleres per a visió subnormal.
- Dispositius electrònics per a baixa visió
- Magnificadors, videolupes.
- Programes especials para PC, programes lectors.
- Telescopis i lupes de mà.
- Apps per a mòbil; llanterna, VoiceOver, etc, etc.
Comunitat Stargardt a la UE modifica
L'Associació D.O.C.E. (Discapacitats Altres Cecs d'Espanya) va proposar a RareConnect l'organització d'una comunitat internacional per posar en contacte a pacients, amics i simpatitzants de Stargardt amb la finalitat d'estar informats sobre els últims avanços i possibles teràpies quan existeixi guareix.
Vegeu també modifica
Referències modifica
- ↑ Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations Jose ́ -Alain Sahel
- ↑ Orphanet: Stargardt disease. Consultat el 3 d'abril de 2013
- ↑ 3,0 3,1 «Stargardt disease : Definition(s) from the Unified Medical Language System ® Diseases Database». [Consulta: 5 febrer 2018].[Enllaç no actiu]
- ↑ Rosa María Coco Martín: Fundus flavimaculatus i malaltia de Stargardt. Consultat el 3 d'abril de 2013